SR-TIGET ex vivo and in vivo lentiviral gene therapy aims to target the CNS, building on MLD success and expanding toward new therapeutic strategies.
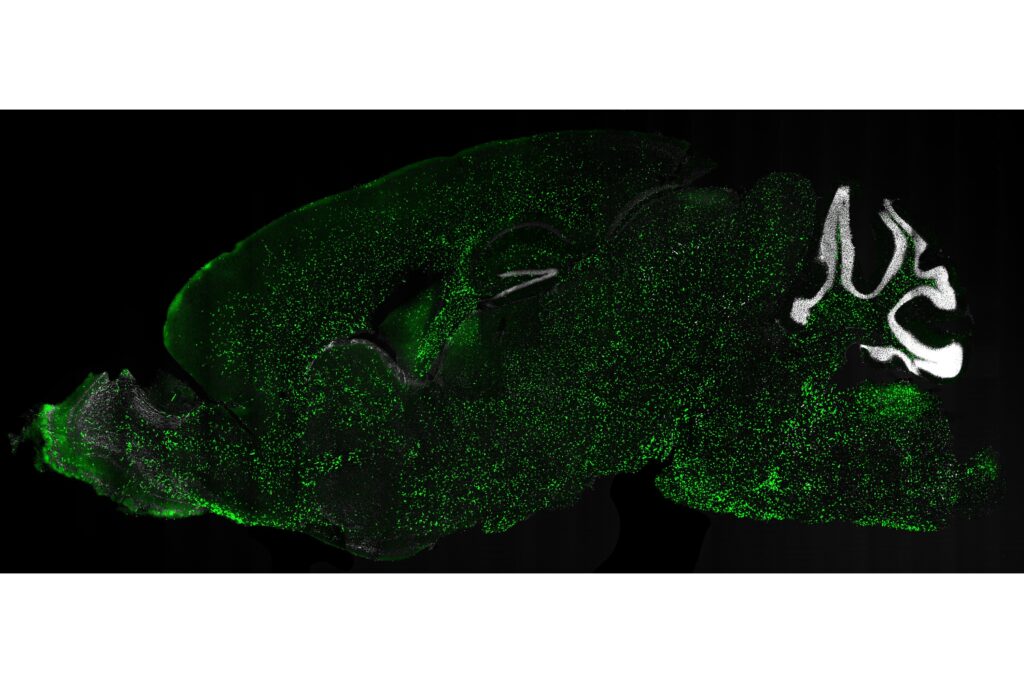
Research into ex vivo and in vivo gene therapy using lentiviral vectors for conditions affecting the central nervous system (CNS) represents a key frontier for the treatment of severe disorders, including rare and ultra-rare diseases.
Within this context, researchers at SR-TIGET are studying and developing increasingly targeted approaches to treat diseases that involve the CNS, with the aim of being among the first to enter clinical testing for in vivo lentiviral gene therapy directed to this target organ.
Work on ex vivo gene therapy based on hematopoietic stem cells (HSC-GT) modified with lentiviral vectors has already led to the development of the first gene therapy medicinal product for metachromatic leukodystrophy (MLD)—a rare genetic lysosomal storage disorder with a neurological phenotype—approved by the EMA in 2020 and by the FDA in 2024.
Building on this milestone, research is moving forward on multiple fronts: on the one hand, towards a deeper understanding of the enzymatic cross-correction mechanisms that underpin clinical benefit; on the other, towards the development of new approaches that could extend treatment opportunities to a growing number of patients and diseases. For other neurodegenerative conditions, SR-TIGET is also exploring alternative delivery strategies as well as gene editing technologies.
Neurological correction in metachromatic leukodystrophy
Metachromatic leukodystrophy (MLD) is a rare neurodegenerative disease caused by the accumulation of toxic substances in the nervous system—sulfatides, which become harmful to myelin-producing cells when present in excess due to a deficiency of the enzyme arylsulfatase A (ARSA). In these conditions, the challenge is not only to identify the most effective strategy to compensate for the missing enzyme, but also to ensure that the enzyme actually reaches the nervous system, including the brain, an organ that is biologically complex and difficult to treat.
The CNS represents a particularly challenging therapeutic target for both conventional drugs and gene therapy. Several biological barriers hinder intervention—most notably the blood–brain barrier, which restricts the passage of molecules and cells from the bloodstream into neural tissue. For a gene and cell therapy strategy to be effective, modified cells must therefore be able to cross these barriers, engraft in the brain and become long-term residents, ensuring sustained and high levels of functional enzyme within nervous tissue. If the enzyme is present only in the bloodstream, it cannot effectively cross these barriers—or does so only minimally—failing to reach sufficient concentrations in the central and peripheral nervous systems.

“The promise of gene therapy is not necessarily to cure the disease, but to change its course if intervention occurs in the right way and at the right time. The goal is to deliver the enzyme where it is truly needed, overcoming biological barriers that normally prevent access to the brain” explains Angela Gritti, Associate Professor of Human Histology at Vita-Salute San Raffaele University and Group Leader of a SR-TIGET research unit dedicated to developing gene therapies for genetic neurodegenerative diseases.
To address this challenge, researchers chose to use autologous blood stem cells collected from the patient and reinfused after genetic engineering through lentiviral vectors. Once the corrected cells engraft in the hematopoietic niche, they differentiate into myeloid cells and migrate throughout the body, including into nervous tissues.
The advantages of stem cell transplantation lie in their ability to self-renew, to persist potentially throughout the patient’s lifetime, and therefore to provide a continuous and stable source of genetically corrected myeloid cells. These cells can cross biological barriers, reach neural tissue and produce high levels of enzyme, exceeding the physiological levels typically obtained through conventional allogeneic transplantation, which has more limited efficacy.
“Transplanting stem cells engineered to produce very high levels of ARSA enables a strong overproduction of the enzyme, particularly in their progeny cells,” says Vasco Meneghini, Senior Researcher (Project Leader) in the SR-TIGET research unit led by Angela Gritti.

Cross-correction through high levels of ARSA expression
This treatment enables the myeloid progeny derived from transplanted stem cells to infiltrate different tissues and contribute stably to the resident myeloid population, including within the central nervous system, where these cells differentiate into macrophages and microglia.
A key feature of this gene therapy approach is the ability of the modified cells to produce enzyme levels higher than physiological ones, which are typically achieved with conventional allogeneic transplantation. Physiological levels of enzyme expression are generally insufficient to fully correct the biochemical defect and are therefore inadequate to effectively counteract disease progression.
Because engineered cells act as high-level enzyme producers, they continuously synthesise and release large amounts of ARSA, which are subsequently taken up by surrounding diseased neural cells through receptor-mediated uptake in a process known as cross-correction. Once inside the affected cells, the enzyme restores their lysosomal degradative activity, enabling the clearance of toxic substrates that have accumulated over time.
“In this ex vivo gene therapy approach, the neural cells themselves are not genetically modified. Instead, they benefit from an indirect correction by recapturing the enzyme released by the engineered, metabolically competent myeloid cells” explains Angela Gritti.
Since a portion of the enzyme is used by the producing cell itself and only part of it is secreted, the amount available for therapeutic action depends directly on the overall level of production: the more enzyme the transplanted engineered cell synthesises, the greater the amount available for correcting neighbouring cells.
“The higher the production, the greater the release, and therefore the larger the fraction available to correct the cells affected by the disease,” concludes Vasco Meneghini.
Two routes to reach the brain: ex vivo and in vivo
For conditions affecting the CNS, such as leukodystrophies and lysosomal storage disorders, therapeutic strategies are not limited to a single approach. In recent years, research has been moving along two distinct but complementary paths: ex vivo, based on the patient’s own hematopoietic stem cells (HSC-GT), and in vivo, which targets the brain tissue directly.
The ex vivo approach: increasing biological efficacy and developing a platform
The current focus of studies using the ex vivo strategy is to increase the biological efficacy of treatment. In these diseases, the amount of enzyme available is a crucial factor: the more enzyme delivered to cells, the greater the possibility of reducing toxic substrate accumulation. For this reason, research is addressing several aspects simultaneously—enhancing enzyme production, improving its secretion, and promoting cross-correction.
Alongside these molecular and functional improvements, however, a fundamental biological limitation remains: the time required for modified cells to reach the brain. After transplantation, the cells must migrate from the bone marrow, where HSC engraft, and repopulate the myeloid compartment of the central nervous system, a process that may take several months in patients.
This is precisely where clinical challenges emerge, particularly in the most aggressive infantile forms of disease.
“It is also important to promote myeloid repopulation of the nervous system by transplanted cells, a process that may require several months. This becomes a major issue in infantile forms, or more generally in very rapid and aggressive diseases: the therapy works, but there is often not enough time for it to act effectively” explains Angela Gritti.
The ex vivo strategy is not limited to metachromatic leukodystrophy. Several diseases belonging to the same family share a common underlying mechanism.
“For years we have been studying not only metachromatic leukodystrophy, but also globoid cell leukodystrophy and another class of lysosomal diseases that are not leukodystrophies—GM2 gangliosidoses,” explains Angela Gritti.
At the root of these conditions lies the same biological principle: an enzymatic defect leading to the accumulation of undegraded material that becomes toxic to cells. In GM2 gangliosidoses, for example, the accumulation primarily affects neurons and therefore represents a form of primary neurodegeneration, whereas in leukodystrophies, the disease may involve additional organs. Nevertheless, the pathogenic mechanism remains the same: a missing enzyme and a substrate that progressively accumulates, disrupting cellular function.
This shared biological basis has encouraged researchers to develop the ex vivo strategy as a true platform.
“The idea is to use the same vector and the same expression cassette for several related diseases, allowing certain safety and toxicity assays to be reused instead of repeating them every time. This would make the process more efficient without compromising safety standards, which remain the priority during the early phases of clinical testing” says Gritti.
The move towards more standardised therapeutic platforms is not limited to research groups. Regulatory authorities are also working to make the evaluation process more efficient for therapies targeting categories of diseases with shared biological characteristics.
The in vivo approach: intervening directly in the brain
Alongside the ex vivo strategy, a second approach has been explored by SR-TIGET scientists to target the CNS, namely in vivo gene therapy. In this case, the lentiviral vector is not used to modify stem cells outside the body but is instead injected directly into the brain parenchyma.
This means that neuronal cells themselves are genetically modified, becoming capable of producing high levels of the therapeutic protein. Once corrected, these cells begin secreting the enzyme, which remains available for cross-correction of neighbouring cells. In addition, part of the released enzyme enters the cerebrospinal fluid, where it can diffuse and reach a broader population of diseased cells.
The main advantage of this strategy is speed. Unlike the ex vivo approach—where transplanted cells require months to migrate and repopulate the brain—in vivo delivery can generate high enzymatic levels in the CNS within a few days, which then remain stable over time. This rapid onset of action becomes particularly important when disease progression is fast.
However, the limitations are equally clear: the therapeutic effect remains largely confined to the CNS. The enzyme produced in the brain does not significantly reach other compartments of the body so the peripheral nervous system and other organs remain untreated.
For this reason, the approach is not conceived as a universal solution but rather as a targeted therapeutic option.
“For metachromatic leukodystrophy, we have considered this strategy primarily for patients with later disease onset, slower progression and less aggressive involvement of other organs. Early infantile forms are extremely aggressive and multisystemic. In those cases, the in vivo approach would not be sufficient because it does not correct the peripheral system or other organs” explains Angela Gritti.
Even in milder disease presentations, however, some peripheral involvement may still occur, although less pronounced. Patient selection becomes a crucial step. The goal is to identify the cohort in which correcting the central nervous system can deliver the greatest clinical benefit.
Two strategies with different objectives
Ex vivo and in vivo approaches are not strictly alternative solutions; rather, they address different clinical needs. The ex vivo strategy aims for systemic correction, using genetically modified hematopoietic stem cells that, over time, also contribute to correcting the central nervous system. The in vivo approach, by contrast, intervenes directly within brain tissue and prioritises the rapid expression of the therapeutic enzyme in the most critical compartment.
The choice between these strategies therefore depends on the specific characteristics of the disease: age of onset, speed of progression and the involvement of organs beyond the central nervous system. In early-onset multisystemic forms, a strategy capable of acting across multiple tissues may be required. In later-onset forms, where neurological damage represents the primary clinical problem, rapid intervention in the brain may be more relevant.
In this sense, the two strategies belong to the same therapeutic framework: both aim to restore the missing enzyme, but through different mechanisms and timelines.
Gene editing: a different strategy for dominant genetic diseases
The approaches described so far fall within the category of gene transfer, meaning strategies that introduce one or more functional copies of a gene to compensate for the absence of an enzyme. Gene editing, however, follows a different logic and may be applied to dominant genetic diseases, where the problem is not the absence of a protein but the presence of a mutated and toxic protein.
Gene editing encompasses a set of techniques that allow precise modifications of DNA, adding, removing or replacing specific sequences through enzymes that function as molecular “scissors and glue”.
One example for which this approach is under investigation at SR-TIGET is Alexander disease, a rare and progressive leukodystrophy of the central nervous system caused by mutations in the GFAP gene. These mutations lead to the formation of aggregates of the GFAP protein, which render astrocytes dysfunctional and ultimately cause demyelination. In this context, the therapeutic strategy does not involve adding a missing gene, but rather modifying the existing one.
Two options are currently being explored: silencing the mutated protein or correcting the mutation by inserting the correct genetic sequence.
“We initially attempted to silence both alleles, because this approach could potentially treat all mutations. At the same time, we are developing strategies to correct only the mutated allele and restore physiological levels of expression” explains Vasco Meneghini.
The development of gene editing presents specific challenges, both in the choice of editing enzyme and in the method of delivery.
The tools currently used for gene correction include enzymes such as Cas9 nucleases and related variants, which act directly on DNA. However, these enzymes originate from bacteria and may therefore trigger immune responses. In addition, there is a risk of genotoxicity if editing occurs in genomic regions other than the intended target.
For this reason, researchers are investigating delivery strategies that limit the duration of exposure to editing enzymes, for example through lipid nanoparticles or viral-like particles, as well as editing approaches that do not entail modifications of the DNA sequence, such as epigenetic editing. The aim is to achieve transient expression of the editing enzyme, thereby reducing both immunological risks and the likelihood of off-target genomic modifications.
Sustaining research: Fondazione Telethon and other funding sources
The development of these and other advanced therapies depends on the continuity of funding.
In this research field, the direct involvement of patient associations is fundamental—not only for financial support but also because they represent a stable and informed partner throughout the entire development pathway.
“Fondazione Telethon is our main reference point, but we also receive funding from European and American patient associations. Additional support can come from the European Union and from the Italian Ministry of Health. Combining multiple funding sources is essential to ensure continuity of research” says Angela Gritti.
Ultimately, progress depends not only on scientific innovation, but also on the ability to integrate robust data, appropriate experimental models, regulatory dialogue and sustained financial support. It is within this balance that research can gradually evolve into real therapeutic opportunities for patients.